Center Highlights
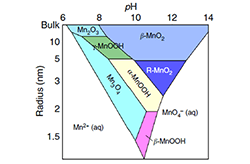
Non-Equilibrium Crystallization Pathways of Manganese Oxides in Aqueous Solution
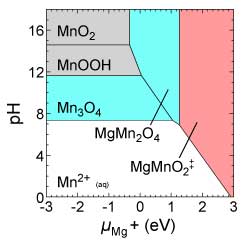
Revealing the metastable energy landscape of Pourbaix diagrams helps to rationalize multistage crystallization pathways of transition-metal oxides from solution.

A Map of the Inorganic Ternary Metal Nitrides
Constructed first comprehensive stability map of inorganic ternary metal nitrides to understand chemical origins of stability and to predict new nitrides, along with corresponding synthetic routes to realize them.
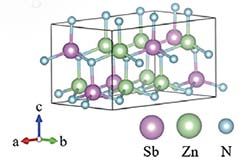
Discovery of a Metastable Photoactive Semiconductor Zn2SbN3
Zn2SbN3 was theoretically predicted and experimentally synthesized—the first Sb-based nitride and a photoactive semiconductor.
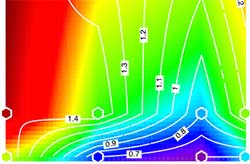
Ternary Nitride Semiconductors in the Rocksalt Crystal Structure
Discovered new class of MgxTM1-xN (TM = Ti, Zr, Hf, Nb) nitrides that bridges properties and structures found in common nitride materials families.
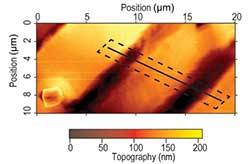
The Existence and Impact of Persistent Ferroelectric Domains in MAPbI3
Methylammonium lead iodide (MAPbI3) exhibits exceptional photovoltaic performance. But a significant controversy remains over the existence and impact of ferroelectricity on the photovoltaic response. We confirm ferroelectricity in MAPbI3 single crystals and demonstrate mediation of the electronic response by ferroelectric domain engineering.
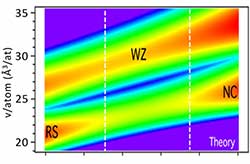
Negative-Pressure Polymorphs via Heterostructural Alloying
For some heterostructural alloys, a metastable low-density polymorph can be the preferred structure for non-phase-separated intermediate compositions. For the MnSe-MnTe system, where this effect was predicted theoretically, the low-density metastable wurtzite phase was successfully synthesized using non-equilibrium thin-film growth.
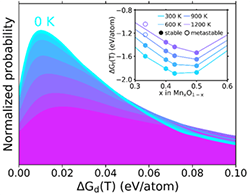
Physical Descriptor for the Gibbs Energy of Inorganic Crystalline Solids and Temperature-Dependent Materials Chemistry December 2018
Developed a machine-learned descriptor enabling the chemically accurate high-throughput prediction of temperature-dependent thermodynamics (<40 meV/at MAE).
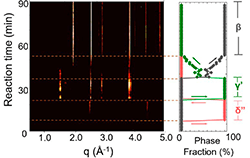
Understanding Crystallization Pathways in MnO2 Polymorphs June 2018
Demonstrated predictive synthesis of MnO2 polymorphs based on principle of remnant metastability—that synthesizable metastable phases nucleate under thermodynamic conditions where they were once the lowest free-energy phase, and then grow into conditions where they are metastable. Using in-situ X-ray diffraction measurements, we verify that this framework can predict which phases are more likely to form as well as the progression order of phases.
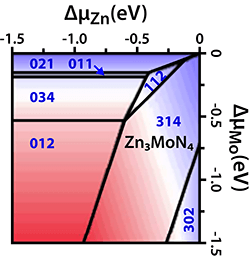
Materials Design Using Redox-Mediated Stabilization (May 2018)
New, stable and metastable Zn-Mo-N alloys with wurtzite-derived crystal structure were theoretically predicted and experimentally synthesized. A broad range of properties—from insulating and transparent Zn3MoN4 to conductive and absorptive ZnMoN2—is realized by tuning the composition.
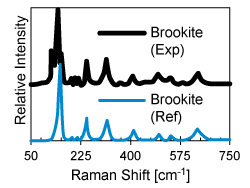
Amorphous Precursors: A Route to Polymorph Synthesis (January 2018)
Using theory-guided synthesis, we have synthesized TiO2 thin films with greater than 80% brookite fraction without substrate templating or the presence of helper ions via solid-phase crystallization of amorphous precursors.
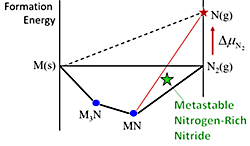
Thermodynamic Routes to Novel Metastable Nitrogen-Rich Nitrides (December 2017)
We formulate how reactive nitrogen precursors can stabilize metastable nitrogen-rich nitrides during synthesis. With this synthesis strategy, we use DFT to predict 22 new nitrogen-rich binary nitrides stabilizable under ΔµN2 = +1 eV/N.
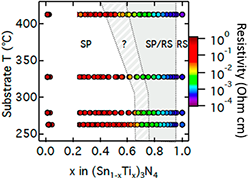
Design of Metastable Tin Titanium Nitride Semiconductor Alloys (November 2017)
We predicted and synthesized new mixed-metal nitride alloys with improved optoelectronic properties. Specifically, we synthesized metastable (Sn,Ti)3N4 spinels using non-equilibrium synthesis. These (Sn,Ti)3N4 alloys have lower hole effectives masses and better transport than Sn3N4.
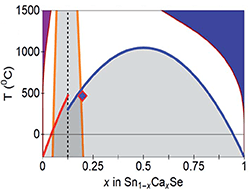
Heterostructural Alloying — A Design Tool to Improve Functionality (October 2017)
Metastable (SnCa)Se with improved thermoelectric functionality was realized by combining theory-guided alloying with non-equilibrium growth.
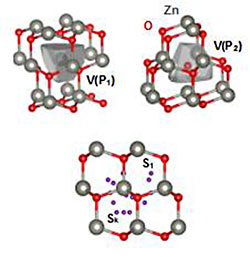
A Framework for Automating Point-Defect Calculations (September 2017)
We completed and rigorously validated an open-source Python framework to automate first-principles point-defect calculations.
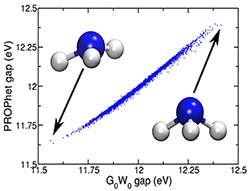
Framework for Coupling Machine Learning and Ab Initio Approaches (June 2017)
Developed computational framework for using neural networks to learn analytical potentials, non-linear density functionals, and structure-property relationships based on high-accuracy ab initio data. Demonstrated use of neural networks to create easily evaluated local DFT charge-density functionals for range of properties for model gas-phase system, NH3.
Phase Selection in MnO2 Structures During Aqueous Growth (May 2017)
We rationalized the synthesis of all common MnO2 polymorphs by forming off-stoichiometric intermediates during aqueous synthesis. We also derived general rules governing off-stoichiometric polymorph stabilization by alkali ions and hydration applicable to transition metal oxide chemistries.
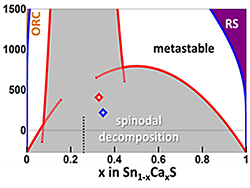
Novel Phase Diagram Behavior in Heterostructural Alloys (April 2017)
We discovered wide metastable regions in the phase diagram of heterostructural alloys. The materials lie above the phase-separated free-energy minimum, but are stable against spinodal composition fluctuations.

ALD Oxides for Higher Performance Power Transistors (February 2017)
Successfully grew single-crystalline (Mg,Ca)O films epitaxially on InAlN transistors with unprecedentedly low density of defects and electron traps at the oxide/semiconductor interface.
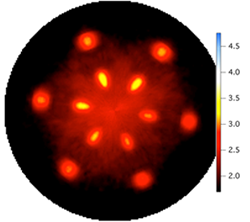
Structure Property Relationships in Wide-Gap Gallium Oxide Thin Films (January 2017)
Successfully determined the synthesis space (substrate temperature, oxygen partial pressure, substrate selection) for the targeted growth of β-Ga2O3 thin films through theory-guided experiments. Grew high-quality, oriented β-Ga2O3 films.
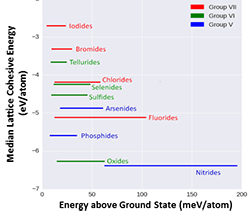
The Thermodynamic Scale of Inorganic Crystalline Metastability (Dec 2016)
Revealed the thermodynamic landscape of inorganic crystalline metastability. Proposed principle of Remnant Metastability: "Observable metastable phases are remnants of thermodynamic conditions where they were once the lowest free-energy phase."
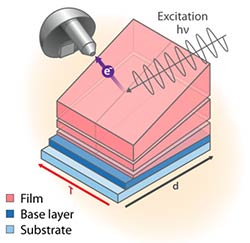
Accelerated in-situ Photo-Electron Spectroscopy for Materials by Design (Nov 2016)
Characterized the interfacial electronic band alignment using new high-throughput measurements by coupling spatially resolved photoelectron spectroscopy (PES) mapping with combinatorially deposited crossed-gradient thin-film samples.
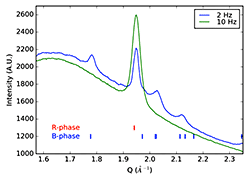
Local Amorphous Structure Controls Polymorph Formation (October 2016)
Used grazing incidence X-ray scattering measurements to identify local structural differences in amorphous thin-film precursors that subsequently determine the polymorph formed upon crystallization.
Epitaxial Polymorph Stabilization through a Computational Approach to Substrate Selection (May 2016)
Developed a computational framework combining calculations of formation energy, elastic strain energy, and topological lattice matching to guide substrate selection for epitaxial materials growth.
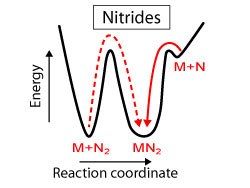
Design of Nitride Semiconductors for Solar Energy Conversion (April 2016)
We provide a new perspective on the promising properties, unexplored chemistry, and metastable character of nitride semiconductors for solar energy conversion.
MnO2 Polymorph Energetics — A Unique Theoretical Challenge (March 2016)
Through a collaboration with the Center for Computational Design of Functional Layered Materials EFRC, we have established that the new, non-empirical, SCAN (Strongly Constrained and Appropriately Normed Semilocal Density Functional) exchange-correlation functional for density functional theory provides a uniquely accurate first-principles model of polymorph energetics and properties.
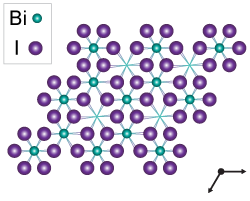
Bismuth Triiodide (BiI3) – A Candidate Photovoltaic Absorber (December 2015)
We identified BiI3 as a candidate photovoltaic absorber using computational design criteria based on the methyl ammonium lead iodide perovskites. Initial experiments demonstrate room-temperature photoluminescence with application-relevant lifetimes.
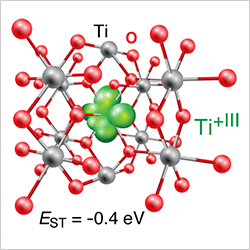
Transition Metal Oxide Semiconductors (Sept 2015)
Comprehensive assessment of semiconducting transition metal oxides using unbiased electronic structure calculations with integrated treatment of s, p, and d electrons.
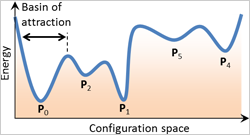
Predicting Polymorphism in Partially Ionic Solids (Aug 2015)
We developed a theoretical approach to search for new and realizable metastable polymorphs in ionic systems.
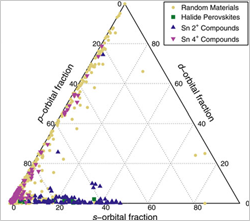
Identifying Defect-Tolerant, High-Lifetime Semiconductors (May 2015)
The key role that band-edge orbital character has on defect tolerance (gained from MAPbX3 perovskites) underlies a new joint data-mining and theory approach to screen materials for long minority-carrier lifetimes, which is a critical photovoltaic absorber property.
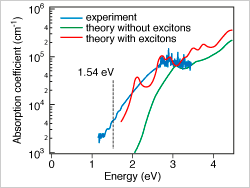
Semiconducting Metastable Sn3N4 and IV3N4 Polymorphs
Thin films of the metastable spinel γ-Sn3N4 were synthesized by sputtering and characterized for semiconducting properties, such as absorption spectra, electrical transport, ionization potential, and minority-carrier diffusion length.
The Center for Next Generation of Materials Design: Mission and Research Plan
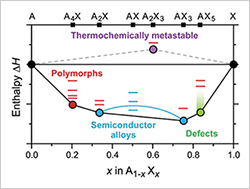
The mission of the Center for Next Generation of Materials Design is to dramatically transform the discovery of functional energy materials through multiple-property search, incorporation of metastable materials into predictive design, and the development of theory to guide materials synthesis.
Incorporating Metastable Polymorphs into Materials by Design
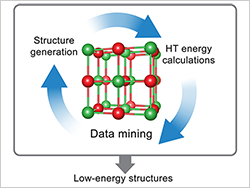
The Center for Next Generation of Materials Design is creating a high-throughput computational tool based on first-principles theory to predict the formation energy of polymorphs, including new unknown structures.
Design of Semiconducting Tetrahedral Mn1-xZnxO Alloys
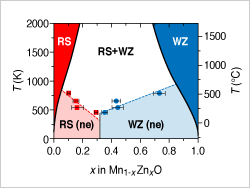
The Center for Next Generation of Materials Design is designing a novel semiconducting transition metal oxide alloy with absorption in the visible and with favorable electron and hole transport properties.